海南德赢·(VWIN)进出口贸易有限公司
Hainan Cuirun Import and Export Trade Co. LTD

分布式注册是欧盟药品注册的两种次要路子,如含有新活性成分的药品、用于严沉疾病的药品、生物手艺药品等。这些药品需要通过EMA进行集中审评,一旦获得上市许可,便能够正在所有欧友邦上市。这种注册路子的劣势正在于可以或许确保药品正在整个欧盟范畴内的快速上市和同一监管。比拟之下,分布式注册法式则愈加矫捷,合用于不正在地方化注册范畴内的药品。申请人能够选择向一个或多个国提交上市申请,由这些国的NCA进行审评。若是药品正在某个国获得上市许可,其他国正在审核后能够认可该决定,并颁布本国的上市许可。这种注册路子的劣势正在于可以或许充实操纵列国的监管资本和专业学问,同时确保药品正在欧盟范畴内的普遍上市。
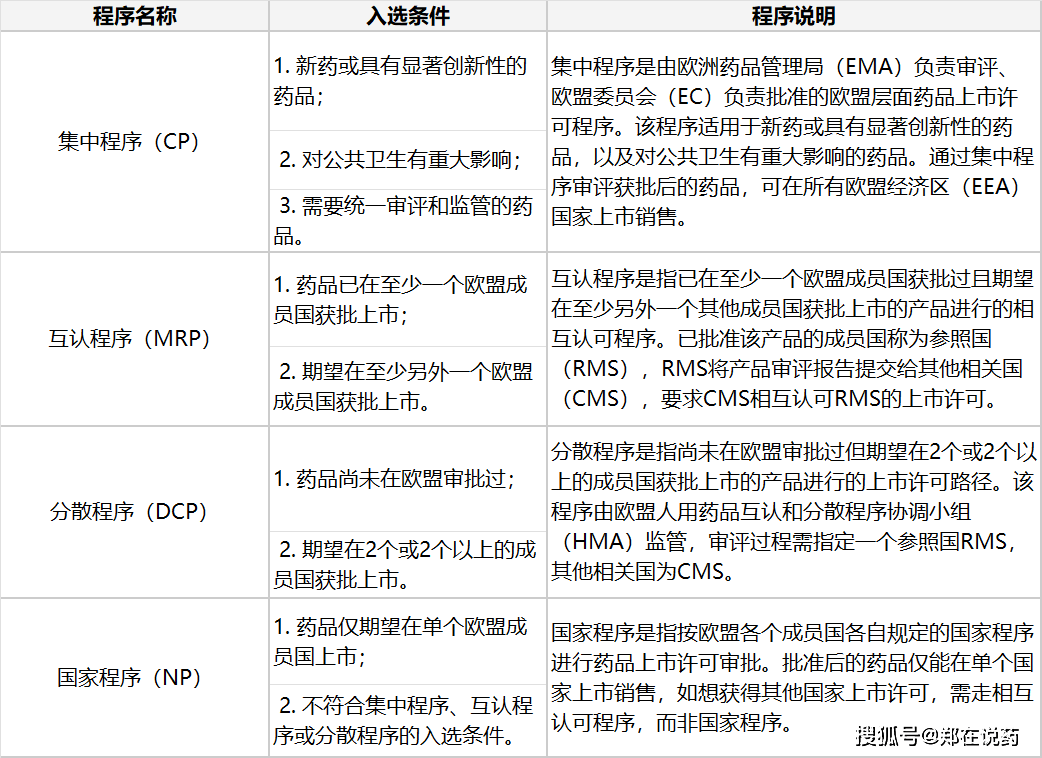
集中法式(Centralized Procedure)、互认法式(Mutual Recognition Procedure, MRP)、分离法式(Decentralized Procedure, DCP)和国度法式(National Procedure)。
合用于CP法式的药品凡是包罗基于先辈生物手艺(如沉组DNA手艺、基因编码节制表达手艺)开辟的药物,正在欧盟境内未上市的含有新活性成分的药物(出格是用于医治癌症、艾滋病、神经退化疾病等严沉疾病的药物),以及孤儿药(即风行病学发病率极低的药物)。
则专注于生物成品的监督工做,生物成品包罗疫苗、血液成品、基因医治产物等。CBER正在生物成品注册中的职责包罗审评生物成品的平安性、无效性和制制质量等方面。审评流程凡是包罗申请提交、材料审查、现场查抄和审评决策等阶段。正在审评过程中,CBER会分析考虑生物成品的临床试验数据、制制工艺、质量节制尺度以及标签和利用申明等消息。取CDER雷同,CBER的审评尺度也基于科学准绳、律例要乞降公共好处等要素。若是生物成品合适平安性和无效性的尺度,CBER将核准其上市发卖,并对其进行持续的监管和监测。
做为欧盟药品监管的焦点计心情构,其的成立源于欧洲经济配合体(EEC)的相关律例,自1995年起正式受理欧盟列国人用和兽用药品的上市申请。其总部位于荷兰,前身为欧洲药批评价局(EMEA),跟着英国脱欧而进行了搬家。EMA的职责笼盖了欧盟范畴内所有药品的科学评价、监视办理和平安性监测,确保药品的平安、无效和高质量。
申报DCP法式时,申请人需要预备取CP法式类似的申报材料,并按照预定的时间表提交给参照国。参照国正在收到申报材料后,将担任协调审评工做,并取其他相关国进行沟通交换。
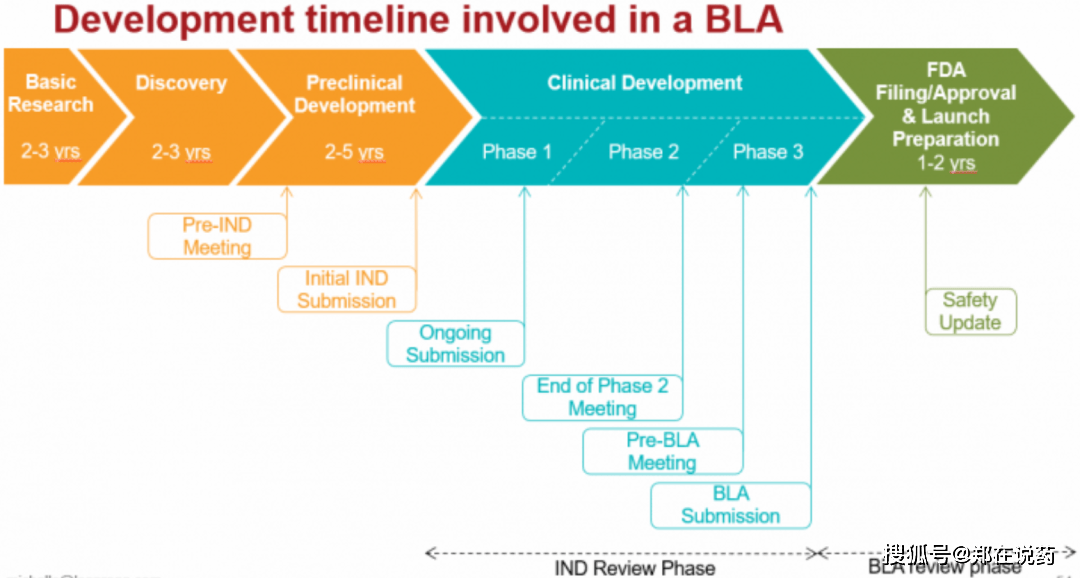
I期临床试验次要评估新药正在健康意愿者中的平安性和耐受性,确定药物的初步平安剂量范畴;II期临床试验正在患有特定疾病的患者中评估新药的无效性和平安性,进一步确定药物的疗效和最佳剂量;III期临床试验则进一步扩大患者样本量,验证新药的临床效益和平安性,并取现有疗法进行比力;IV期临床试验是新药上市后的监测阶段,旨正在评估新药正在现实利用中的持久平安性和疗效。
新药申请(NDA)、简单新药申请(ANDA)以及生物成品许可申请(BLA)等多个方面。NDA。
具体来说,模块1要求供给产物特征概要、专家材料、风险评价以及孤儿药品市场独有性的相关材料,这些正在中国尚未做为需要内容要求供给。模块2至4正在分歧地域是不异的,但模块1则具有地域性。此外,欧盟对药品标签和包拆仿单的格局和内容有严酷要求,需要供给平面设想样稿、现实印刷样品及其可读性测试申明。

中国的药品注册流程履历了多次,旨正在缩短新药上市周期,提高审批效率,同时确保药品的平安性取无效性。企业需提交详尽的申报材料,履历、受理、审评、审批等多个环节,曲至最终获得上市许可。美国FDA的注册流程则以其矫捷性取立异性著称,如快速通道、冲破性疗法认定等机制,为医治严沉或稀有疾病的药物供给了加快审批的绿色通道。欧洲EMA则通过集中审批法式,实现了国间的快速互认,降低了企业的注册成本取时间成本。

NMPA的次要使命包罗:担任药品、医疗器械和化妆品的尺度办理,组织制定和发布国度药典等尺度;担任注册办理,制定注册办理轨制,严酷审评审批;担任质量办理,制定研制和出产质量办理规范并监视实施;担任上市后风险办理,监测、评价和措置不良反映和不良事务;担任执业药师资历准入办理,依法查处违法行为;同时,NMPA还担任对交际流取合做,参取国际监管法则和尺度的制定,并指点省、自治区、曲辖市药品监视办理部分的工做。
正在组织架构上,NMPA设有分析和规划财政司、政策律例司、人事司、科技和国际合做司(港澳台办公室)等分析机构,以及药品监视办理司、药品注册办理司(中药平易近族药监视办理司)、医疗器械监视办理司、医疗器械注册办理司、化妆品监视办理司等营业机构。
注册分类的差别则间接反映了三地对于药品立异的激励策略取市场需求。中国近年来加大了对立异药的支撑力度,通过优先审评审批、专利链接等政策,激发了企业研发立异药的热情。美国FDA则通过孤儿药认定、快速通道等机制,为医治稀有病、严沉疾病的药物供给了更多上市机遇。欧洲EMA则通过集中审批法式取国间的互认机制,推进了立异药的快速畅通取普遍使用。
中国、欧盟和美国均采用了药品注册通用手艺文件(CTD)做为同一的申报文件格局。CTD将申报材料分为五个模块,包罗地域性行政办理材料、研究内容概要和综述、质量研究演讲、非临床研究演讲以及临床研究演讲。这种格局沉视研究成果表述的具体化和规范化,使得评审机构可以或许更清晰地领会申报药物的特征、平安性和无效性。
申报MRP法式时,申请人需要预备细致的申报材料,包罗已获批国的上市许可文件、药品的质量、平安性和无效性数据以及药品标签等消息。这些材料将提交给参照国(即已获批国的药品审批机构)进行审评。参照国正在收到申报材料后,将对其进行审评,并预备审评演讲。
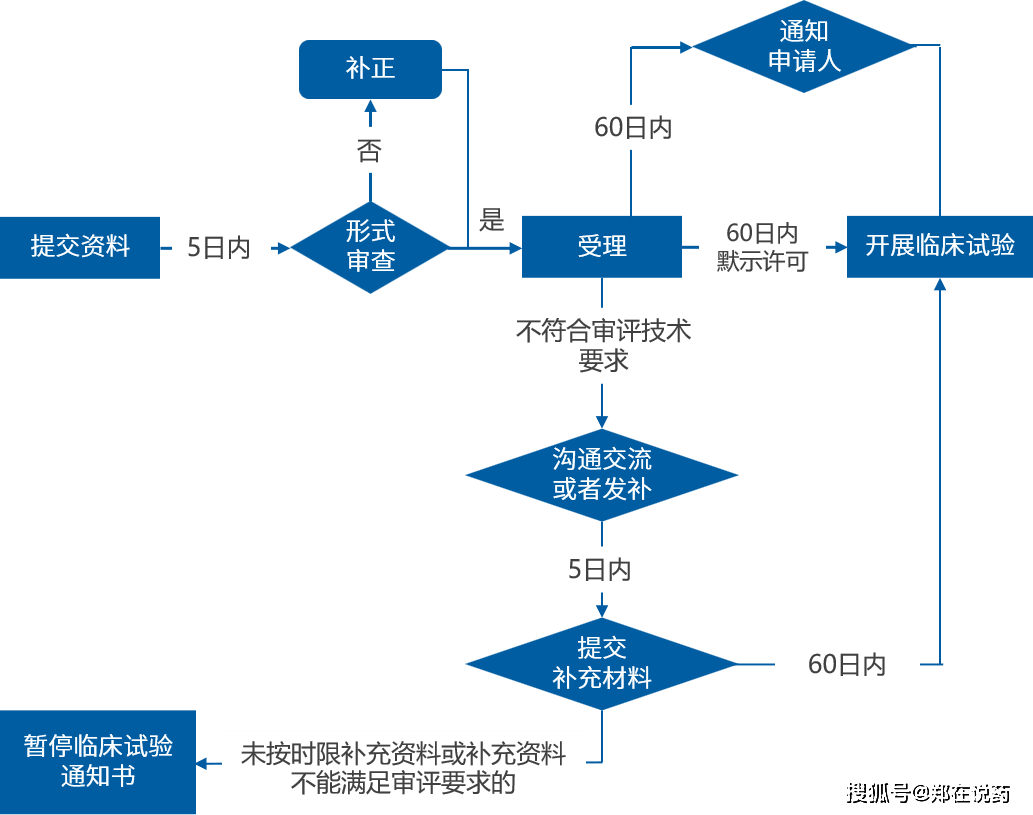
欧洲药品注册流程是确保药品可以或许正在欧洲市场上平安、无效、地发卖和利用的主要环节。这一过程涉及多个法式,此中集中注册法式(CP)!
正在审评过程中,NMPA会对申报材料的实正在性、完整性和规范性进行审核,并要求企业供给额外的数据或消息。若是进口药合适平安性和无效性的尺度,NMPA将颁布进口药品注册证书,答应进口药正在中国市场发卖。
欧盟的药品注册分类包罗完整申报(新药)、简化申报(仿制药)以及其他特殊类型药品。此外,欧盟还有一些特殊类型的药品分类,如复方(固定组方)药物、改良型仿制药以及知情同意药等。复方药物是由两个以上现有原料药构成的药物,用于特殊的顺应症和用处。改良型仿制药则是正在参比制剂的根本长进行规格、给药路子或顺应症等方面的优化。知情同意药则是申请的药品取相关国已许可的药品素质类似,而且原药品上市许可持有者同意将原药品的毒理、药理和/或临床材料用于所述申报药品的评价。
中美欧三个地域的药品注册机构事实有哪些?它们的注册流程又是若何的?这些流程之间又存正在着如何的差别呢?继续阅读,带你一探事实!
防止用生物成品和医治用生物成品两大类。防止用生物成品次要是疫苗,而医治用生物成品则包罗用于人类疾病医治的生物成品,如卵白质、多肽及其衍生物等。中药注册分类。
此中,优先审评机制下,FDA许诺正在6个月内完成审评,这比尺度审评周期缩短了近一半。此外,若新药合适加快核准路子(Accelerated Approval Pathway)的前提,即药物可以或许处理未满脚的医疗需求且其临床好处可以或许正在晚期临床试验中获得,那么FDA能够正在基于替代起点或两头临床起点的根本上核准药物上市,进一步缩短上市时间。

FDA对简单新药申请的审评尺度次要包罗仿制药的生物等效性、质量可控性以及取原研药的分歧性。审评时限凡是为6个月,但具体时间可能因申请类型、复杂程度和审评资本等要素而有所分歧。若是仿制药合适相关尺度和要求,FDA将颁布简单新药核准函,答应仿制药正在美国市场发卖。
仿制药注册和进口药注册三大类,每一类都有其特定的要乞降流程。新药注册流程涵盖了从临床前研究降临床研究,再到新药申请和审批的全过程。临床前研究是新药研发的起始阶段,次要方针是评估药物的平安性、药代动力学特征以及初步的疗效。这一阶段包罗药物的合成、制剂研究、药效学研究、药代动力学研究、毒理学研究等。
生物成品,如疫苗、血液成品、基因医治产物等。生物成品许可申请的预备阶段需要完成全面的临床前研究和临床试验,以证明生物成品的平安性和无效性。提交阶段,申请人需将完整的生物成品许可申请材料提交给FDA的生物成品审评取研究核心(CBER)。2。3 欧洲药品注册流程。
正在审评过程中,EMA可能会多次取申请人进行沟通,以解答审评过程中的疑问,确保审评的精确性和高效性。审评周期凡是为210天,但具体时间可能因申请类型、复杂程度和审评资本等要素而有所分歧。若是药品合适平安性和无效性的尺度,答应药品正在整个欧洲经济区(EEA)国度上市发卖。

国度药品监视办理局(NMPA)是中国药品监管的焦点计心情构,其本能机能涵盖了药品(包罗中药、平易近族药)、医疗器械和化妆品的平安监视办理。NMPA不只担任制定和监视实施相关法令律例、政策规划以及部分规章,还担任研究订定激励新手艺新产物的办理取办事政策。
正在欧盟药品注册中,NCA取EMA慎密协做,配合确保药品的平安性和无效性。具体来说,NCA担任提交国度法式的药品上市申请,进行药品平安,并施行GMP(优良出产规范)、GCP(优良临床实践)等相关查抄。同时,NCA还参取EMA的科学委员会工做,为EMA供给科学和帮帮。这种协做机制确保了欧盟药品注册工做的成功进行,以及药品监管的分歧性和高效性。
此中,集中法式是EMA最次要的审评法式,合用于具有立异性或医治严沉疾病的新药。集中法式的审评周期一般为210天,即从EMA收到完整申请材料之日起算,包罗90天的初步评估期和120天的细致评估期。然而,考虑到申请人可能需要弥补材料或EMA可能需要进一步评估,现实审评时间可能会更长。
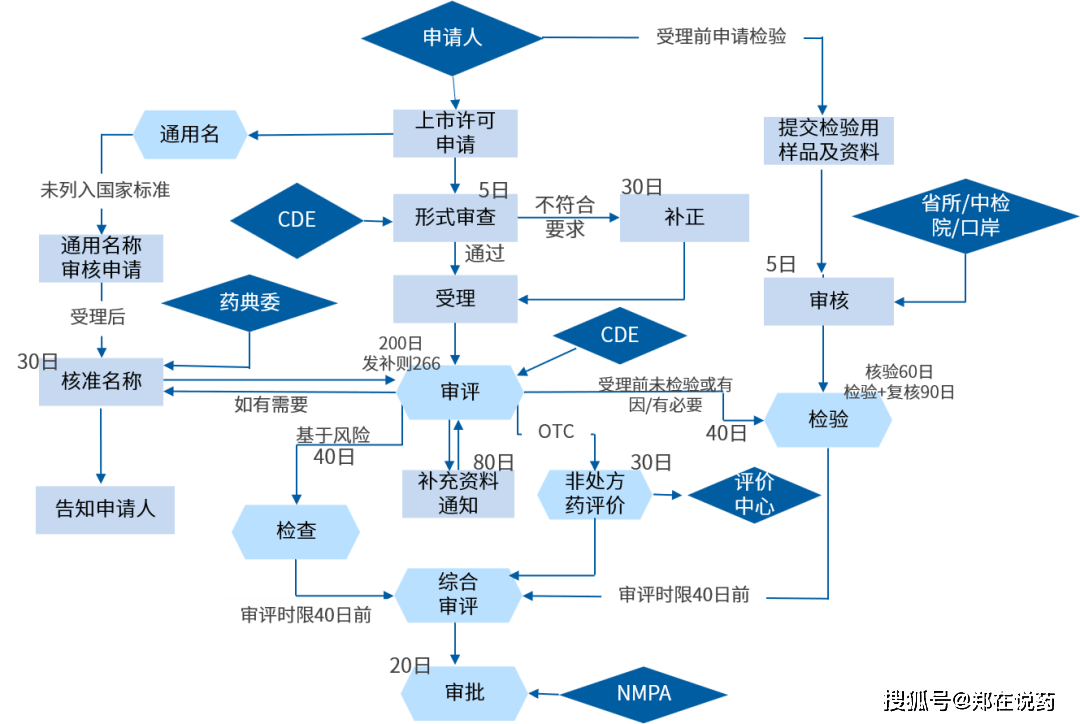
CDE的审评时限按照分歧类型的注册申请而有所分歧。例如,对于新药注册申请,CDE需要正在的时间内完成审评工做,并给出能否核准的决定。对于需要弥补材料的申请,CDE会一次性奉告申请人需要弥补的内容,并正在收到弥补材料后继续审评。CDE还设有沟通交换机制,为申请人供给手艺,避免弯,加速药物研发和上市进度。同时,CDE还担任药品注册查验和注册核查工做,确保药品注册申请的线 美国药品注册机构?。
省级药品监视办理部分正在药品注册方面,担任辖区内药品的监视办理工做,包罗贯彻实施国度和省级药品监视办理法令律例,草拟相关处所性律例、规章草案,拟定相关规划、政策并监视实施。它们还担任药品的行政监视和手艺监视,贯彻施行国度药典等药品尺度,以及药品研制、出产、运营、利用质量办理规范。正在药品注册办理上,省级药监部分取NMPA协同工做,确保药品注册流程的成功进行。此外,省级药监部分还担任成立药品不良反映监测系统,开展监测和措置工做,以及奉行国度根基药物目次,共同实施国度根基药物轨制。
美国食物药品监视办理局(FDA)对新药注册的审评时限相对较短,这得益于其高效的审评流程和完美的律例系统。一般环境下,新药申请(NDA)的审评周期约为10至12个月,但这并非绝对,具体时间取决于药物的复杂性、申请人提交材料的完整性和FDA的工做负荷。

药品审评核心(CDE)是国度药品监视办理局的曲属机构,其次要职责是担任药品注册申请的审评工做。CDE的审评流程凡是包罗申请受理、材料审查、沟通交换、审评审批等环节。正在审评过程中,CDE会根据相关律例、政策和手艺要求,对药品的平安性、无效性和质量可控性进行全面评估。CDE的审评尺度严酷遵照国度药典等药品尺度,确保药品注册申请的科学性、规范性和性。
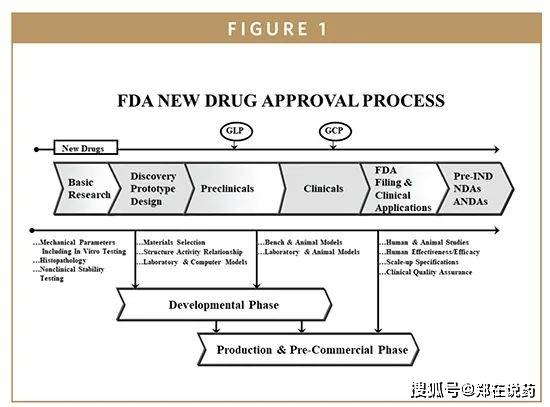
FDA正在收到新药申请后,将启动审评流程,审评过程包罗手艺审评、现场调查以及多次取申请人的议。手艺审评次要关心新药的平安性、无效性、质量可控性以及风险评估。现场调查则是对申请人的出产设备进行现场查抄,以确保其合适GMP(优良出产规范)要求。议则是为领会答审评过程中的疑问,确保审评的精确性和高效性。按照律例,FDA凡是应正在6个月内完成新药申请的审评,但具体时间可能因申请类型、复杂程度和审评资本等要素而有所分歧。
自2015年以来,NMPA启动了一系列办法,包罗加速审评速度、优化审评流程、加强取国际监管机构的合做取交换等。这些办法使得新药注册时限大幅缩短,对于合适优先审评前提的新药,如具有较着临床劣势的立异药、儿童用药、稀有病用药等,NMPA许诺正在130个工做日内完成审评,这比尺度审评周期缩短了近一半。
全新化学实体、新实体或显著改良现有疗法的新药。正在预备阶段,申请人需要完成全面的临床前研究和临床试验,以证明新药的平安性和无效性。临床前研究涉及药理学、毒理学、而临床试验则分为I、II、III期,逐渐正在健康意愿者、特定疾病患者中进行,以评估药物的疗效、平安性和最佳剂量。一旦临床研究和数据收集完成,申请人需要细心预备新药申请材料,这些材料凡是包罗细致的药品仿单、临床研究演讲、制制工艺和质量尺度、非临床研究数据等。新药申请材料往往很是复杂,可能达到数万页以至更多,需要细心组织和拾掇。提交阶段,申请人需将完整的申请材料提交给美国食物药品监视办理局(FDA)的药品审评取研究核心(CDER)。
正在中国,国度药品监视办理局(NMPA)做为药品注册的焦点监管机构,其本能机能涵盖了从新药研发到上市后的全生命周期办理,表现了中国对药品平安取立异的双沉注沉。美国方面,食物药品监视办理局(FDA)以其科学严谨、高效通明的监管系统闻名,不只保障了美国国内药品市场的健康运转,更正在全球药品监管范畴树立了标杆。欧洲药品办理局(EMA)则通过集中审批法式取国间的互认机制,无效推进了欧洲药品市场的同一取繁荣,展示了区域一体化正在药品监管范畴的奇特魅力。
是FDA中担任药品审评的焦点部分,旨正在确保新药的平安性和无效性。审评流程凡是包罗新药申请(NDA)的提交、审评预备、审评实施和审评决策等阶段。正在审评过程中,CDER会分析考虑药品的临床试验数据、制制工艺、质量节制尺度以及标签和利用申明等消息。审评标原则基于科学准绳、律例要乞降公共好处等要素。CDER的审评时限因申请类型和复杂程度而异,但凡是会正在的时间内完成审评工做。若是药品合适平安性和无效性的尺度,CDER将向制药公司颁布核准函,答应其正在美国市场上发卖新药。

正在中国,新药注册现场查抄凡是包罗注册核查、注册查验和飞翔查抄,注册核查次要核实申报材料的实正在性、分歧性以及药品上市贸易化出产前提,查抄药品研制的合规性、数据靠得住性等;注册查验则是对药品样品进行尝试室查验,以验证其能否合适药品注册尺度;飞翔查抄则用于监测现行cGMP(药品出产质量办理规范)和其他要求的持续合规性。

包罗一个办理委员会和多个科学委员会,办理委员会担任EMA的日常办理和决策,而科学委员会则专注于药品的科学评价和监管。具体来说,EMA下设有人用药品委员会(CHMP)、药物鉴戒风险评价委员会(PRAC)、兽用药品委员会(CVMP)等多个科学委员会,它们别离担任分歧范畴的药批评价和监督工做。此外,EMA还设有流程办理和贸易支撑部分、查抄和药物鉴戒部分等多个支撑性部分,以确保EMA可以或许高效、专业地履行其职责。
FDA的焦点构成部门包罗药批评价取研究核心(CDER)、生物成品评价取研究核心(CBER)、医疗器械取放射健康核心(CDRH)、食物平安取使用养分核心(CFSAN)、兽医学核心(CVM)以及烟草产物取尼古丁监管核心(CTP)等。FDA次要担任监管美国国内出产或进口的食物、化妆品、药物、生物制剂、医疗设备和放射产物!

:通过科学评价和监管来保障和提高和动物的健康;严酷对欧盟内的药品上市申请进行审评,确保药品的平安性和无效性;监测药品的平安,通过欧盟的平安监测或药物鉴戒系统来持续性地评估药品的收益和风险均衡;参取特殊议题中的药品保举法式,处理关于药品平安或收益风险均衡的问题;担任实施欧盟的近程消息处置项目,以及激励药品的立异和研发等。
正在中国,申报材料的格局和内容要求则相对复杂。按照《药品注册办理法子》的,申报材料被分为中药取天然药物、化学药品和生物成品三大类别,每个类别下又有多个子类。例如,中药和天然药物项下细分为9类产物,化学药品项下分为6类产物,生物成品项下分为15类产物。分歧的类别有分歧的注册办理模式和申报材料要求。
仿制药注册流程凡是包罗:企业提交仿制药注册申请,包罗仿制药的质量尺度、出产工艺、不变性研究数据、BE试验数据等消息。NMPA将对申请进行审评,包罗对仿制药的质量、平安性和生物等效性的评估。
正在加速审评政策的实施方面,中国近年来加速了新药注册审评速度,推出了优先审评、冲破性医治药物法式、附前提核准法式和出格审批法式等多种加速审评政策。这些政策旨正在缩短新药上市周期,提高患者用药可及性。美国FDA则设立了快速通道、冲破性疗法、优先审评和有前提核准等多种加速审评政策,以激励新药研发和立异。欧盟EMA则通过PRIME通道、加快审评、有前提核准和特例核准等政策,加速新药正在欧盟市场的上市速度。